
21 CFR Part 11: Increased URS Complexity for Software Compliance
Whenever a new instrument is introduced into a pharmaceutical lab, it is normally commissioned through a process of qualification and validation. Depending on a company’s policy, this process is probably based on the most recent USP 1058 or GAMP 5… or a combination of both.
Use of a User Requirements Specification (URS) document is recommended in both of the USP 1058 or GAMP 5 guidance documents. The URS document truly defines the system and is a cornerstone of the validation.
THE IMPORTANCE OF URS AND ROLE IN VALIDATION
PerkinElmer’s validation services team has been active in hundreds of validations across numerous environments, witnessing, first-hand, the evolution of pharmaceutical validation and its change in the approach to creating URS documents.
Prior to the revision of USP 1058, the vast majority of pharmaceutical companies were taking a strict GAMP 5 approach and often encountered a misapplication of the risk-based approach. This resulted in over-engineered validation projects: too many category 3 systems were validated with category 4 rigor.
These validation packages contained several Requirements Documents, e.g. design requirements, configuration requirements, functional requirements, and finally user requirements or sometimes called performance requirements.
Fortunately, these processes have matured over the years and we’ve observed the streamlining of most validation processes. Many companies have consolidated the multiple requirements documents into a single “URS” or “RS” document.
Generally, one would expect that this trend to lead to a URS document. But in contrast, we are hearing from more and more customers who require third-party expert support for their URS documentation. Conveniently for clients, since PerkinElmer is both an instrument company AND service provider, we can package URS support along with the physical instrument sale.
This inclusion is very useful. More often than not, the sections that are most struggled over are those detailing security and the data management lifecycle – in other words, the requirements brought about by the integration of 21 CFR Part 11 into the commissioning and validation process.
But why? 21CFR Part 11 has been around for over 20 years.
HISTORY OF PART 11 and IMPACT ON URS
In the early 1990s, traditional paper-based processes were being replaced by automated approaches. Increasingly, vendors of analytical instrumentation were bringing to market computerized systems. These software driven systems evolved over time – starting with the capability to operate the instrument, but eventually having the ability to process data and manage data completely electronically. Instrument vendors were taking their cues from the wants of the industry – and the industry was seeking to streamline business processes and to go “paperless”.
As this wave of change swept across the industry, large parts of the CFR which governed those traditional processes, the predicate rules, were becoming less aligned with the actual practices of the industry. Key groups who operated within the pharmaceutical industry met with the FDA to determine how the industry would approach these changes and still maintain compliance with the predicate rule.
Early versions of Part 11 were drafted by these groups and the rule was made final, becoming effective on August 20th, 1997. There’s a lot more history beyond this point… but it’s important to understand that, like most regulations, the original rule was not well received by the industry. Complying with the new regulations placed additional burden on everyone – vendors, scientists, quality professionals.
Even today, this burden, and the subsequent industry backlash, has left residue. And that residue is probably most evident in the increasing complexity found within a typical URS document. To explain, the new rule had to be integrated with all systems in the GxP lab including instrumentation. The best way to do this, was to translate the rule into requirements and incorporate into a URS.
DRIVERS OF COMPLEXITY
As stated, Part 11 has been around for over 20 years, and the concept of a URS document even older than that. So why the lasting complexity?
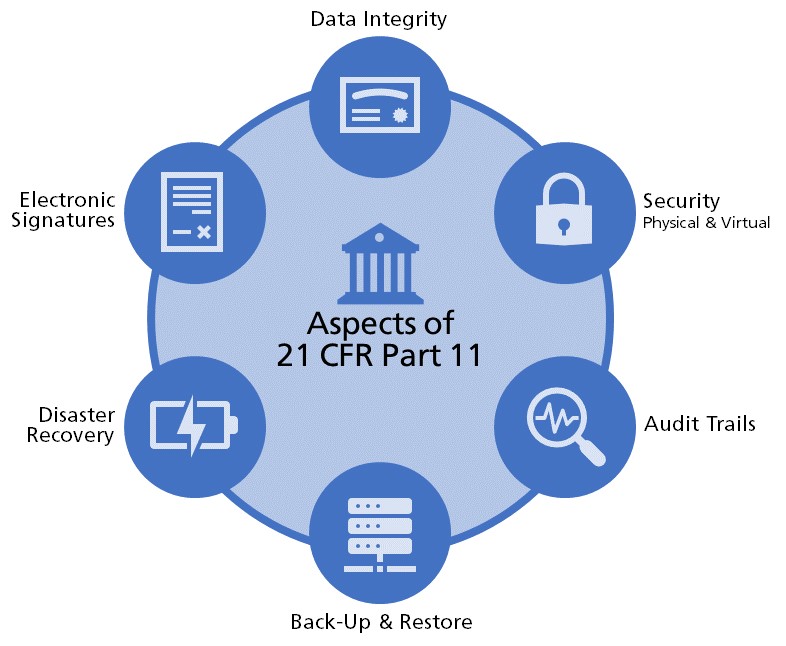
- General misunderstandings of what software should and shouldn’t do:When part 11 gets translated into requirements, it normally is broken up into a few different categories: Security, Data Integrity, Electronic Signatures, Audit Trails, Disaster Recovery, Back-Up and Restore.
- Vendor Software Quality and Capability:This situation has improved in recent years, but the issue was that even though instrument vendors took great strides to design software meant for Part 11 compliance, they often fell short. Two of the primary short falls that I’ve seen were
- Many vendors failed to secure data (it could be deleted, primarily), and;
- audit trails were weak and not up to par with part 11.
In light of no good (from a part 11 perspective) instrument options, the folks tasked with validation had to consider work-arounds and proceduralization. This had a “trickle-up” effect in the validation package, and URS documents were somewhat compromised in that requirements were selectively included and not standardized.
A TWO-STEP PATH FORWARD
- Take a templated approach to URS documents, and standardize those requirements dealing with Part 11 and related topics. Standardize those requirements that are met by company policy. Most requirements except for those dealing with the intended use of the instrument should apply across vendors and instrument families. This approach is enhanced by having a foundation of strong Data Integrity and Data Management policies.
- Use a Specialized Supplier Qualification/Assessment form prior to instrument purchase. This form is for the vendor to fill out and will list your part 11 requirements as well as solicit the part 11 capabilities of the system in question. This mechanism is used to ensure that any new system purchased will be a drop-in fit with your standardized URS and validation approach – again streamlining the validation effort and reducing the need for workarounds. Use of a tool like this is quickly becoming a best practice in the industry.
Overall, by working together, the situation is already trending toward improvement. Instrument vendors are producing more compliant software, and industry knowledge on Part 11 and other Data Integrity topics are improving. In addition, there are actions that can be done to simplify the URS and streamline the overall validation process.